Editor’s note: Seeking Alpha is proud to welcome Asker Mammadov as a new contributing analyst. You can become one too! Share your best investment idea by submitting your article for review to our editors. Get published, earn money, and unlock exclusive SA Premium access. Click here to find out more »
Editor’s note: Seeking Alpha is proud to welcome Asker Mammadov as a new contributing analyst. You can become one too! Share your best investment idea by submitting your article for review to our editors. Get published, earn money, and unlock exclusive SA Premium access. Click here to find out more »
Thesis
With a couple of catalysts approaching in the next year and a half for Prothena Corporation plc’s (NASDAQ:PRTA) late-stage assets, I analyze the medical literature and past clinical trial data to get an idea of what the future holds. I believe these late-stage assets will fail their upcoming catalysts, which in turn will be bearish for the valuation of the stock.
Overview of Prothena’s Pipeline
Prothena currently has 9 assets in development (many in collaboration) within their pipeline that specialize in neurodegenerative and protein misfolding diseases. None of these are approved, which means future cash flows are dependent on the clinical trials being successful for milestone payments or for revenue. Most of the assets are quite early to very early stage. The latest stage drugs are 2 monoclonal antibodies: Birtamimab for AL-Amyloidosis and Prasinezumab for Parkinson’s. These are the assets that are most likely to have an impact on the price of the stock. I believe these late-stage assets will fail their upcoming trials leading to a further drop in the stock price. They also have another phase 2 drug NNC6019 (PRX004) in collaboration with Novo Nordisk A/S (OTCPK:NONOF) (NVO) (however, this is early phase 2, and I do not believe it has too much of an influence on investor sentiment).
Prothena Pipeline (Prothena)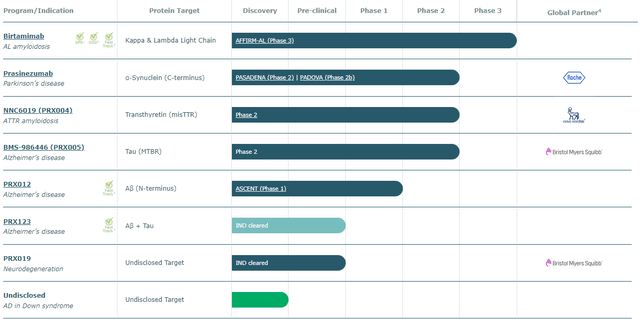
A Look at Prothena’s Financials
According to their latest financial report on the 8th of May, as of March 31, they had around $550 million in cash/equivalents. They received an additional $80 million from a Bristol Myers Squibb Company (BMY) opt-in as of 28 May for clearing an IND for PRX-019. Averaging their last 5 quarters, they have a cash burn rate of approximately $250 million/year ($62.5 million/quarter). That gives them a runway of around 2-2.5 years. The stock has been on a downtrend for almost exactly a year from a high of $78.6 on May 5th, 2023.
Birtamimab: AL-Amyloidosis
Birtamimab is a novel monoclonal antibody therapy directed against AL-Amyloid fibrils in AL-Amyloidosis which leads to dissolution of the fibrils. These fibrils are deposited throughout key organs, of which the heart is the most important, and eventually lead to death. Currently, no immunotherapy treatments against fibrils exist in AL-Amyloidosis. Around 60-120 thousand people are expected to have Stage 4 AL-Amyloidosis throughout the world and >20 thousand in Europe and the US (thus making this a rare disease and Birtamimab an Orphan drug). Birtamimab is wholly owned by Prothena. It is currently in Phase 3 of development. It had previously failed another Phase 3 trial (“VITAL”) and is now awaiting topline results for its Phase 3 “AFFIRM-AL” trial, which is expected between Q4 2024 and Q2 2025.
Prasinezumab: Parkinson’s Disease
Prasinezumab is a monoclonal antibody directed against α-Synuclein in Parkinson’s Disease. Around 1 million people in the U.S. and ~1.2 million in Europe are expected to have P.D. Prasinezumab is being developed in collaboration with Roche. To date, Prothena has received $135 million in milestone payments from their Prasinezumab collaboration with Roche out of a total potential $600 million. The results of Prasinezumab’s phase 2b “PADOVA” trial are expected by the beginning of Q4 2024.
In the sections that follow, I will present a detailed assessment of why I believe these two key candidates – Birtamimab and Prasinezumab – are likely to fail late-stage trials.
Valuation
The 2 trials are essential catalysts for the stock price. The stock price at the time of writing of this article (29 May) is ~$20. With 53.8 million shares outstanding, the pure cash value of the stock, based on cash and cash burn rates, would be 590/53.8, or ~$11 (the 590 is derived from 550 – ~40 (2 months of cash burn since 31 March) + 80 (the BMS opt-in)). That means the market is valuing their entire pipeline at around 20-11=$9/stock (This is, of course, merely an estimate and is liable to change depending on investor sentiment, so one could give or take. For example, right before the $80 million milestone payment 2 days ago by BMS, the stock price of PRTA was still $20, so with those numbers, the pipeline would be valued slightly higher). Logically, the majority of the value of the pipeline lies in the late-stage drugs at this current point in time (as the other drugs are early to very early stage. The only exception to this was 1-2 years back when the Alzheimer’s hype was very strong and Prothena’s very early stage PRX-012 was causing their stock to skyrocket.) which means at the very least, using the numbers above, 9*0.5=$4.5/stock, if not more, of value lies in the upcoming trials providing significant topline results. It also makes sense the majority of this $4.5/stock value is expected from Prasinezumab (perhaps 75% for Prasinezumab, 25% for Birtamimab) as Parkinson’s is a far more prevalent disease than AL-Amyloidosis which results in far greater potential future royalty revenues as well as the fact that they get milestone payments (they also don’t need to worry about development costs as much) whereas with Birtamimab they do not get any milestone payments and the entire onus is on Prothena for commercialization. Thus, if it were to be revealed that Birtamimab and Prasinezumab failed in their end goals, the stock could potentially overall lose up to ~$4.5-6 (plus additional cash burns that would have elapsed in those timeframes).
Risks & Strengths to this thesis
Given my thesis is bearish for the stock (based on the medical science as written below), any sudden positive news like new potential collaborations with upfront payments, as we saw very recently with Bristol Myers Squibb, within the next 2 quarters could cause a rise in the stock price. (Although these would be small payments, it is still something that could sway investor sentiment).
The bigger risks to my thesis would be encouraging topline results from the late-stage studies. If Prasinezumab were to succeed, they would most likely get a milestone payment of ~$100 million (as mentioned above, they have $465 million in potential milestone payments left out of a total $600 million) but keep in mind this is still phase 2b and Prasinezumab would still have to pass Phase 3 for regulatory/market approval after which they would get 70/30 split on revenue in the U.S. and double-digit royalties in ex-U.S sales. It is quite tricky to forecast how much revenue would come from sales after approval for Prasinezumab as neurodegenerative immunotherapies have had a very rocky past few years (so we don’t even have a starting point from which we can model a forecast). To give you an idea, Biogen Inc.’s (BIIB) Leqembi for Alzheimer’s was supposed to be the first success story but the phase 3 results were very mediocre and showed quite dangerous side effects leading to a very slow adoption in the medical community even though it was FDA approved. The closest thing to any form of true success was Eli Lilly’s Donanemab for Alzheimer’s, with a list price of ~$26,500/year (same as Leqembi), which showed very good results in its phase 3 trial (something that Prasinezumab has yet to do) and seemed like it was pretty much set up for FDA approval until they delayed it due to serious safety concerns as well. Currently, P.D is projected to have a total addressable market of ~ 12 billion by 2030 (and that isn’t even with the consideration of immunotherapy treatments having a heavy contribution). Despite there being no guidance from Prothena or Roche for how much revenue potential could be expected from Prasinezumab, we can derive a rough estimate using $26,500 as a reference point, a 50% penetration into the market at peak, the U.S. and European population as the target demographic and assuming a 30% rebate on the list price for the US and 20% for Europe. For the US population, 1 mil * $26,500 * 0.5 * 0.7 = $9.275 billion (at peak) of revenue potential. For the EU population, 1 mil * $26,500 * 0.5 (drugs are on average half price in Europe) * 0.8 = $5.3 billion. Therefore, a total revenue potential of $9.275 + 5.3 = $14.575 billion
The same principles would apply to Birtamimab if it were to be successful in its phase 3 trial. Add on top of the fact that Birtamimab would not have any competition as it is significantly ahead of everyone in development and that all profits would go to Prothena since Birtamimab is wholly owned. However, AL Amyloidosis is quite a rare disease, and Stage 4 is even more, so potential future cash flows would be smaller than Prasinezumab. Approval of Birtamimab could see up to a revenue potential of (assuming a cost of around $200,000 (since AL-Amyloidosis Mayo Stage 4 is a rare disease), a minimum patient population of 20,000 throughout U.S. and Europe, and a rebate of 20%) we would get an approximate revenue potential of $200,000 * 20000 * 0.8 =$3.2 billion
Strengths for my bearish thesis in addition to my hypothesis of the trials failing. While Prasinezumab might be the only remaining late-stage P.D immunotherapy trying to obtain approval (after discontinuation of Cinpanemab by Biogen), it still has a lot of competition from a lot of other molecular class drugs. Relating to concepts in the previous paragraph, successful trials, and even approval would not be enough to warrant adoption by the medical community. The effect of Prasinezumab would have to be quite clinically significant (not just statistically significant) otherwise it risks becoming another Leqembi/Aduhelm (the same applies to Birtamimab), especially in the face of other competition.
Now that we have the financial analysis out of the way, I will dive into the medical science behind why I believe the upcoming late-stage assets will be unsuccessful in their trials.
Birtamimab’s success is an unlikely story
Even though Birtamimab is a first-in-class experimental amyloid depleter which leads the pack, I do not believe Birtamimab will be successful in increasing the primary endpoint of time to all-cause mortality within Mayo Stage 4 patients relative to Placebo in the upcoming “AFFIRM-AL” trial. The primary reasons for this are:
1) A post-hoc analysis finding that wasn’t corrected for in the previous Phase 3 VITAL Trial was used as an endpoint for AFFIRM-AL.
2) The survival benefit being restricted to the most severe stage but not stage 3 in the post-hoc group doesn’t make sense, given that the mechanism of action of Birtamimab should have an easier task of dealing with stage 3 patients from a pathophysiological point of view. The investigators of VITAL point out in the “Discussion” section that “It is unlikely that this study would have been able to detect a difference in survival between treatment groups in patients with Mayo stages I-III AL amyloidosis without a considerably longer duration of treatment, given the reported median survival for patients with Mayo stage I, II, and III AL amyloidosis of ∼94, 40, and 14 months, respectively”. However, if we look at supplemental Figure 2, we will find the original primary endpoint Kaplan Meier chart for the VITAL trial which compared the survival curve of all Birtamimab patients against all placebo patients (that is not just Mayo Stage IV but all stages). This chart shows that the trial had actually lasted a total of 34 months.
Kaplan Meier estimate of primary endpoint for all stage groups in “VITAL” trial (American Society of Hematology)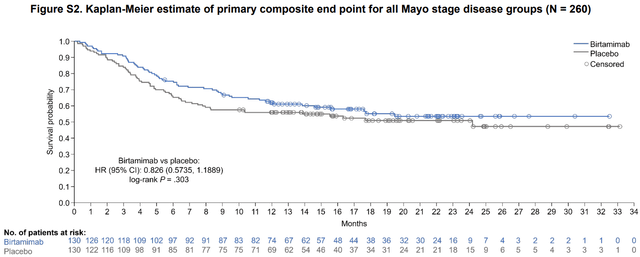
Supplemental Figure 2
Considering that the median survival times of the different stages quoted by the investigators, for Stages 1, 2, 3 are 94, 40, and 14 months respectively, I could understand the idea that there wasn’t enough of a “duration of treatment” for Stage 1 and Stage 2 but not for Stage 3 which leads me to believe that there was no successful effect with Birtamimab in Stage 3 patients which is still quite a severe stage of AL Amyloidosis. Therefore, in the absence of any scientific reasoning for why the effect would be present in stage 4 and not 3 the only thing left to deduce is that it was due to haphazard chance/other factors beyond our understanding.
3) No difference in cardiac organ response biomarkers (Table S6) between Birtamimab and Placebo patients, which has a significant association with increased survival, in the post-hoc stage 4 group and is used in many consensus guidelines for monitoring and prognosing cardiac involvement in AL Amyloidosis.
Cardiac Best response in Post-hoc group in “VITAL” Trial (American Society of Hematology)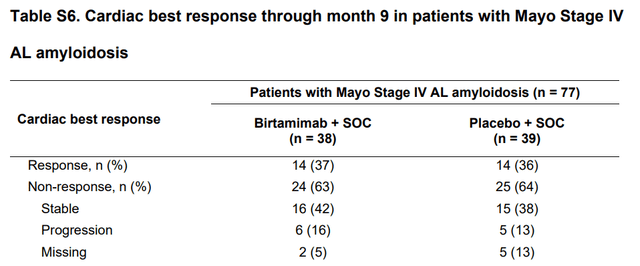
Prothena themselves had carried out earlier Phase I/II trials with Birtamimab (study name “NEOD-001”) whereby they had noted impressive organ responses which prompted them to further pursue Birtamimab.
4) Certain baseline characteristics (variances within “Number of involved organs at baseline”, “Baseline NT-proBNP, pg/mL” “Baseline dFLC”) could explain why placebo did worse than Birtamimab in the stage 4 cohort post-hoc group. Emphasis on the difference in quartile ranges
Baseline characteristics in “VITAL” Trial (American Society of Hematology)



5) Secondary endpoints didn’t show a useful clinical significance
The secondary endpoints the investigators analyzed in the Post-Hoc group in the VITAL trial were:
a) The change in SF-36v2 PCS at month 9 (which is a quality-of-life questionnaire)
b) The change in the 6-Minute Walk Test distance at month 9
The SF-36v2 PCS score change from baseline difference between the two groups of stage 4 patients was shown to be +4.65 in favor of the Birtamimab group, with a P-value of 0.046. The PCS score can have a maximum of 100. Therefore, a difference of 4.65 is not that much. The P-value is also barely significant. But the real problem with using this secondary endpoint as a measure of the success of Birtamimab is that the questions in the survey are subjective, as opposed to say the 6MWT (which is objective).
Moving onto the “6-Minute Walk Test”. The difference between placebo and Birtamimab in stage 4 patients was 36 meters in favor of the Birtamimab group, with a P-value of 0.022. While this is a much more objective test relative to the PCS survey, a difference of 36 meters isn’t much. Let’s assume the effect actually was statistically significant. Would a 36m difference in heart function actually be considered clinically significant? I will leave the interpretation up to the reader.
6) Survival in AL Amyloidosis is much more multifactorial than just AL Amyloid Deposits in organs.
A quick recap on the pathological mechanism of AL Amyloidosis. Defective plasma cells produce misfolded light chains. These misfolded light chains can exist in 1 of 3 configurations at a given time. Starting with monomeric forms which can then clump into oligomeric forms (Soluble Aggregates) which can further clump together to result in amyloid fibrils within organs (insoluble deposits)
All three configurations have been shown to impair cardiac function in preclinical models (in vitro and in vivo) where Monomers/Soluble Aggregates are directly toxic to cardiomyocytes (cells) through internalization whereas amyloid fibrils cause metabolic dysfunction through extracellular means as well as compromising tissue architecture (Interestingly enough, in another type of amyloidosis known as ATTR patients can present with as much amyloid in their hearts as AL and yet, their survival is far greater than AL Amyloidosis which is believed to be due to the TTR amyloid protein being far less disruptive/toxic). Furthermore, pre-clinical models show that there can be combinations (For instance, toxic monomers/oligomers but not very metabolically disruptive fibrils or vice versa). What makes the situation even more complicated is that AL Amyloidosis is actually a heterogeneous disease. While researchers might classify a certain group of people as all having AL Amyloidosis, every individual’s misfolded light chain is unique. This is due to the fact that the light chains play an important role in forming antibodies against many different types of antigens (foreign substances) and in order to be able to do that they need to be able to adopt many different types of configurations under normal conditions (something that’s known as V(D)J gene recombination). Add on top of that amyloidogenic mutations within the gene segments of the light chain and one can see how complex it can get. Because of this heterogeneity, certain patients can have far more damaging light chains or light chains that are far more prone to fibril formation.
Birtamimab can get rid of the insoluble deposits through macrophage-induced phagocytosis and is also claimed to be able to neutralize soluble aggregates (unfortunately, there is no published data in the medical literature on the efficacy of Birtamimab neutralizing soluble aggregates. (see here, Palladini et al. under section 5 “Amyloid-depleting mechanism of action of Birtamimab”). However, the monomer forms are not neutralized by Birtamimab as it requires an epitope(site) that is revealed when it aggregates with other light chains. This is left to the S.O.C chemotherapy to handle by eliminating the plasma clones that produce it.
Taking into account what was stated in the last 2 paragraphs, we can Imagine scenarios where Birtamimab would have a meaningful impact on survival and others where the survival of the patient is at the mercy of the chemo successfully eliminating the plasma cell. For instance, a patient with a toxic monomer but not a toxic oligomeric/fibrillar form wouldn’t gain too much of a survival benefit from Birtamimab and would depend mainly on the chemotherapy being successful (which it isn’t most of the time) as opposed to say a patient that has monomeric forms that aren’t toxic, whereas their oligomeric/fibrillar forms are. Since current diagnostic methods are not efficient (either time or cost-efficient) enough to quickly classify patients and their subtypes of AL Amyloidosis, clinical trial investigators are essentially blinded in figuring out which AL patients would most benefit from Birtamimab.
In addition to this, we are dealing with the most fragile patients in Stage 4. These patients are usually the ones that have been exposed to the amyloids the longest and also usually have the most deposits in their organs, which is also a function of time. At a certain point throughout the disease, the damage becomes permanent and organ dysfunction can no longer be reversed. So even if Birtamimab actually were to be successful in removing the deposits, in many (maybe even most) cases it would be too late in improving survival.
Due to their highly fragile state, the effect of chemotherapy cannot be ignored either. On the one hand, chemo might help with the elimination of the LC-producing plasma cells, while on the other hand, it could be contributing to cardiovascular adverse events itself. Thus, this could potentially offset the effects of Birtamimab on survival. Furthermore, chemotherapy is known to have a myelosuppressive effect (bone marrow immune cell generation is hampered). The mechanism of Birtamimab is dependent on macrophages to clear the deposits and circulating soluble aggregates. A lot of the chemotherapies used in AL Amyloidosis have examples of inducing myelosuppression. While the specific effect they have on the monocyte lineage (the lineage that gives rise to macrophages, which are important for the Birtamimabs Mechanism) is not well researched, it is something to keep in mind.
7) A significance level of 0.1 for the AFFIRM-AL trial increases the risks of a false-positive.
Taking into account all of the previous points, I believe there is a fairly decent probability that Birtamimab will fail its upcoming trial, which would be bearish for the stock.
Prasinezumab is facing a steep hill
The upcoming phase 2b trial “PADOVA” has “Time to Confirmed Motor Progression Event” as a primary endpoint where the inclusion criteria favor “rapidly progressive” Parkinson’s Disease
Investigators claim that Prasinezumab has more success in preventing motor progression in “rapidly progressive” P.D according to a post-hoc exploratory analysis and an Open-Label Extension of the PASADENA study (where an external observation group, the PPMI cohort, was used as a control group).
However, I believe this trial will most likely fail due to the following reasons:
1) The SPECT Scan showed no difference between treatment groups in dopaminergic activity in PASADENA which is the entire basis behind motor progression in P.D since a gradual neuronal loss will lead to less dopaminergic neuronal density(hence weaker SPECT scan) and more motor symptoms
2) In an extension of the PASADENA study (from 52-104) weeks when the Control patients were transferred to Prasinezumab at Week 52(“Delayed Start Cohort”) their mean change worsened between 52-56 weeks. (Figure Below).
Change in Score on MDS-UPDR Part III from Baseline to Week 104 (New England Journal of Medicine)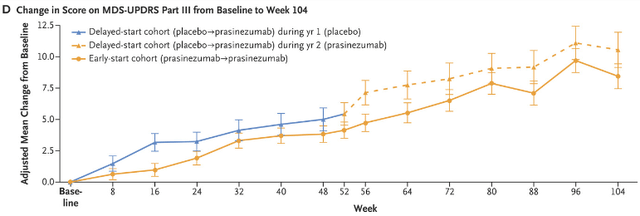
3) Using post-hoc exploratory analysis leads to spurious relations (same problem as Birtamimab above) and using an external cohort in the open label-extension study compromises the randomization aspect of the gold standard of RCT’s (not to mention that this was done after the fact.)
4) Another problem is that Roche/Prothena don’t actually know if Prasinezumab successfully bypasses the human Blood-Brain Barrier because they can’t “…incorporate testing for target engagement because such tests are not well developed.” Antibodies are very large molecules and thus it makes it very difficult for them to actually pass the BBB unless they are engineered for receptor-mediated transport (which is not the case with Prasinezumab).
5) Even if Prasinezumab bypasses the BBB, its goal is to bind to α-Synuclein in the extracellular space and thus prevent it from being internalized by neurons. However, studies have shown that α-Synuclein can propagate through nanotubing mechanisms without ever having passed through the extracellular space, effectively shielding them from binding.
6) The reasoning behind Prasinezumab is that by binding α-Synuclein and preventing its uptake by surrounding neurons, it will prevent neuronal loss. However, a study shows that neuron loss already happens before Braak Stage 3 (when there still is no α-Synuclein within the neurons in the Substantia Nigra) indicating that other factors could be causing neuronal loss.
7) While it is true that extracellular α-Synuclein uptake adds to the burden of neurons, it is most likely dwarfed by the contribution of intracellular mechanisms due to genetic mutations such as SNCA, LRKK2, PINK1, PARK, etc., and environmental factors. These mechanisms aren’t only restricted to neurons but are present in other cells of the CNS as well (microglia and astrocytes) which can convert them into neuroinflammatory phenotypes that promote cell death alongside the burden of α-Synuclein accumulation.
8) In preclinical studies on mice for Prasinezumab (“9E4”) they use transgenic mice models that emphasize α-Synuclein gene overexpression(mThy1-α-syn mice, PDGF-β promoter mice). These models don’t consider lysosomal defective pathways (which are the most common mutations in familial P.D which shares overlap with Sporadic P.D) such as LRKK2 (the most frequent mutation in P.D) or GBA. This could be the reason why most mice were successful in getting rid of α-Synuclein in pre-clinical models with Prasinezumab (because their lysosomal pathways are intact). However, with patients who do have defective lysosomal pathways, α-Synuclein would not get broken down and continue to accumulate within the CNS cells.
9) Cinpanemab, another antibody directed at α-Synuclein in the extracellular space, was unsuccessful in showing a difference between Control and Drug in PART III of MDS-UPDR (however it binds another epitope compared to Prasinezumab, the N-Terminal, and has less affinity to certain types of a-aggregates).
With all of the above points in mind, it seems to me that it would be unlikely for Prasinezumab to succeed, which would be bearish for the valuation of the stock.
Read the full article here
